
Edad al diagnóstico: 3 años
Tiempo hasta el diagnóstico: <1 year
Síntoma clave de diagnóstico: Soplo cardíaco
Médico que realizó el diagnóstico: Cardiólogo pediátrico
Paciente con MPS VI, edad 0 a 17
Descripción1:
Resumen
EL diagnóstico temprano de MPS VI permitió la rápida intervención y tratamiento, que están asociados con mejores resultados clínicos.2
Los síntomas clásicos de MPS —baja estatura, otitis media recurrente, manifestaciones esqueléticas y reumatológicas — junto con el compromiso cardíaco, son indicadores de una consulta inmediata al genetista o centro del metabolismo.2
Inicio de terapia de reemplazo enzimático (TRE)





Descripción1:
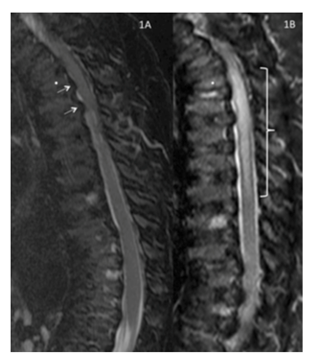
Recuperación de inversión T2 sagital /T1 corta ―imagen ponderada de la columna torácica y lumbar obtenida 7 meses antes de la operación.
Reproducida con permiso de Drummond, Can J Anesth, 2015.
Resumen1
En este caso ilustrativo, la lesión de médula espinal post-operatoria fue desproporcional al grado de estenosis espinal. Este caso es un ejemplo de la exagerada vulnerabilidad de las personas con Morquio A a la compresión y daño de médula espinal.
Los anestesiólogos deben considerar la estenosis espinal subclínica con laxitud de articulación de la columna, además del riesgo de compresión.2
Historia del caso1
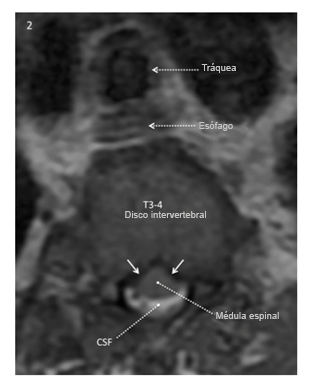
La imagen ponderada axial T2 al nivel del disco intervertebral T3-4 obtenida 7 meses antes de la operación muestra fluido cerebroespinal (blanco) alrededor de la parte posterior de la médula espinal, no de la anterior. La superficie anterior de la médula tiene una configuración en forma de “carpa” en lugar de una configuración suave convexa, lo que refleja presión ventrolateral bilateral en la superficie anterior de la médula (flecha blanca sólida).
Reproducido con permiso de Drummond, Can J Anesth, 2015.
Basado en un estudio proporcionado por el Dr Zakharchuk
Descripción1:
Abreviaturas: 3MSC, escalamiento de 3 minutos; 12MWT, test de caminata de 12 minutos; FEV1, volumen respiratorio forzado en 1 segundo; FVC, capacidad vital forzada; GAG, glicosaminoglicano.
Resumen1
El diagnóstico temprano es fundamental para el inicio de la TRE, cuando se encuentre disponible, y brinda la posibilidad de mejorar los resultados de los pacientes.3-6,b Tal como se demuestra en este caso, la TRE tiene el potencial de mejorar los parámetros clínicos claves, tales como mediciones de resistencia y respiratorias, que pueden ser críticas para la calidad de vida del paciente, el mantenimiento de la ambulación y las actividades de la vida cotidiana.7,8
Historia del caso1
Edad al diagnóstico: 12 años
Tiempo hasta el diagnóstico: 11 años
Síntoma de diagnóstico clave: Retraso motor y del habla
Médico que realizó el diagnóstico: Genetista
La aparición de síntomas neurológicos a la edad de 1 año, seguidos de una rápida progresión del retraso de desarrollo, lleva al diagnóstico de MPS IIIA en 2 hermanas1
Descripción1:
Resumen1
Como se demuestra en este caso, los signos y síntomas pueden progresar rápidamente, especialmente con deterioro neurológico y empeoramiento del retraso en el desarrollo en pacientes con MPS IIIA. En las familias con varios niños que presentan cualquiera de los rasgos indicadores de MPS, debe existir un índice de sospecha particularmente alto.1
A pesar de que el monitoreo y la evaluación continua son críticos, se recomienda la intervención temprana a través de una consulta al genetista y/o centro del metabolismo, en lugar de una detección metabólica usando uGAGs, para agilizar el diagnóstico.1
Historia del caso1
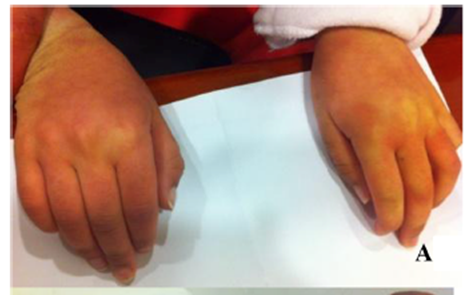
Los exámenes revelaron una descoloración en la piel en pacientes con MPS IIIA.1
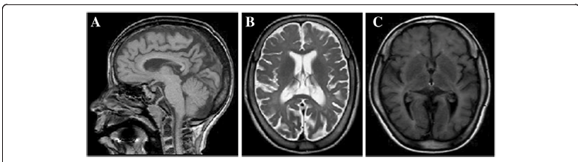
La MRI reveló hipomielinación difusa, afinamiento del cuerpo calloso y atrofia cerebral moderada, síntomas que se desarrollaron con el paso del tiempo.1
References: 1. Data on file. Biomarin Pharmaceutical. 2. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 3. Giugliani R, Lampe C, Guffon N, et al. Natural history and galsulfase treatment in mucopolysaccharidosis VI (MPS VI, Maroteaux-Lamy syndrome)–10-year follow-up of patients who previously participated in an MPS VI Survey Study. Am J Med Genet A. 2014;164A(8):1953-1964. doi:10.1002/ajmg.a.36584.
References: 1. Drummond JC, Krane EJ, Tomatsu S, Theroux MC, Lee RR. Paraplegia after epidural-general anesthesia in a Morquio patient with moderate thoracic spinal stenosis. Can J Anesth. 2015;62(1):45-49. doi:10.1007/s12630-014-0247-1. 2. Spinello CM, Novello LM, Pitino S, et al. Anesthetic management in mucopolysaccharidoses. ISRN Anesthesiol. 2013;2013:1-10. doi:10.1155/2013/791983.
References: 1. Data on file. Biomarin Pharaceutical. 2. Hendriksz CJ, Berger KI, Giugliani R, et al. International guidelines for the management and treatment of Morquio A syndrome. Am J Med Genet Part A. 2014;9999A:1-15. doi:10.1002/ajmg.a.36833. 3. Valayannopoulos V, Nicely H, Harmatz P, Turbeville S. Mucopolysaccharidosis VI. Orphanet J Rare Dis. 2010;5:5. doi:10.1186/1750-1172-5-5. 4. Clarke LA. Pathogenesis of skeletal and connective tissue involvement in the mucopolysaccharidoses: glycosaminoglycan storage is merely the instigator. Rheumatology (Oxford). 2011;50(suppl 5):v13-18. doi:10.1093/rheumatology/ker395. 5. Lehman TJA, Miller N, Norquist B, Underhill L, Keutzer J. Diagnosis of the mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v41-v48. 6. Morishita K, Petty RE. Musculoskeletal manifestations of mucopolysaccharidoses. Rheumatology. 2011;50(suppl 5):v19-v25. doi:10.1093/rheumatology/ker397. 7. Muenzer J. Overview of the mucopolysaccharidoses. Rheumatology. 2011;50:v4-v12. doi:10.1093/rheumatology/ker394. 8. Hendriksz C. Improved diagnostic procedures in attenuated mucopolysaccharidosis. Br J Hosp Med. 2011;72(2):91-95. 9. Muenzer J. Early initiation of enzyme replacement therapy for the mucopolysaccharidoses. Mol Genet Metab. 2014;111(2):63-72. doi:10.1016/j.ymgme.2013.11.015.
Reference: 1. Sharkia R, Mahajnah M, Zalan A, Sourlis C, Bauer P, Schöls L. Sanfilippo type A: new clinical manifestations and neuro-imaging findings in patients from the same family in Israel: a case report. J Med Case Rep. 2014;8:78. doi:10.1186/1752-1947-8-78.


